
上海细胞库
人源细胞系| 稳转细胞系| 基因敲除株| 基因点突变细胞株| 基因过表达细胞株| 重组细胞系| 猪的细胞系| 马细胞系| 兔的细胞系| 犬的细胞系| 山羊的细胞系| 鱼的细胞系| 猴的细胞系| 仓鼠的细胞系| 狗的细胞系| 牛的细胞| 大鼠细胞系| 小鼠细胞系| 其他细胞系|
人源细胞系| 稳转细胞系| 基因敲除株| 基因点突变细胞株| 基因过表达细胞株| 重组细胞系| 猪的细胞系| 马细胞系| 兔的细胞系| 犬的细胞系| 山羊的细胞系| 鱼的细胞系| 猴的细胞系| 仓鼠的细胞系| 狗的细胞系| 牛的细胞| 大鼠细胞系| 小鼠细胞系| 其他细胞系|

| 规格 | 价格 | 库存 |
|---|---|---|
| 50ul | ¥ 980 | 200 |
| 100ul | ¥ 1680 | 200 |
| 200ul | ¥ 2480 | 200 |
| 中文名称 | 成纤维细胞生长因子受体2抗体 |
| 别 名 | KGFR; KSAM; Bacteria expressed kinase; BEK; BEK fibroblast growth factor receptor; BFR 1; BFR1; CD 332; CD332; CD332 antigen; CEK 3; CEK3; CFD 1; CFD1; Craniofacial dysostosis 1; Crouzon syndrome; ECT 1; ECT 1; ECT1; FGF receptor; FGFR 2; FGFR-2; Fgfr2; FGFR2_HUMAN; Fibroblast growth factor receptor 2; Hydroxyaryl protein kinase; Hydroxyaryl protein kinase; Jackson Weiss syndrome; JWS; JWS antibody K SAM; K sam protein; K sam protein; K-sam ; Keratinocyte growth factor receptor 2; Keratinocyte growth factor receptor; Pfeiffer syndrome; Protein tyrosine kinase receptor like 14; TK14; TK25; Tyrosylprotein kinase; Tyrosylprotein kinase. |
| 研究领域 | 肿瘤 心血管 免疫学 神经生物学 信号转导 干细胞 生长因子和激素 激酶和磷酸酶 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 交叉反应 | Human, Mouse, (predicted: Rat, ) |
| 产品应用 | ELISA=1:500-1000 IHC-P=1:100-500 IHC-F=1:100-500 Flow-Cyt=1μg/Test ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复) not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 89kDa |
| 细胞定位 | 细胞膜 分泌型蛋白 |
| 性 状 | Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human FGFR2:21-120/821 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 储 存 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| 保存条件 | Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. |
| PubMed | PubMed |
| 产品介绍 | Fibroblast growth factors (FGFs) are members of a large family of structurally related polypeptides that are potent physiological regulators of growth and differentiation for a wide variety of cells of mesodermal, ectodermal and endodermal origin. Four genes encoding for high affinity cell surface FGF receptors (FGFRs) have been identified: FGFR1, FGFR2, FGFR3 and FGFR4. FGFRs are members of the tyrosine kinase family of growth factor receptors. FGFR2 is highly expressed in developing human tissues including the brain, choroids plexus, lung etc. Alternative names: Bacteria expressed kinase; BEK; BFR 1; BFR1; CD 332; CD332; CD332 antigen; CEK 3; CEK3; CFD 1; CFD1; Craniofacial dysostosis 1; Crouzon syndrome; ECT 1; ECT1; FGFR 2; Fibroblast growth factor receptor 2; Hydroxyaryl protein kinase; Jackson Weiss syndrome; JWS; K SAM; K sam protein; Keratinocyte growth factor receptor 2; Keratinocyte growth factor receptor; KGFR; KSAM; Pfeiffer syndrome; Protein tyrosine kinase receptor like 14; TK14; TK25; Tyrosylprotein kinase. Function: Tyrosine-protein kinase that acts as cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation, migration and apoptosis, and in the regulation of embryonic development. Required for normal embryonic patterning, trophoblast function, limb bud development, lung morphogenesis, osteogenesis and skin development. Plays an essential role in the regulation of osteoblast differentiation, proliferation and apoptosis, and is required for normal skeleton development. Promotes cell proliferation in keratinocytes and immature osteoblasts, but promotes apoptosis in differentiated osteoblasts. Phosphorylates PLCG1, FRS2 and PAK4. Ligand binding leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. Phosphorylation of FRS2 triggers recruitment of GRB2, GAB1, PIK3R1 and SOS1, and mediates activation of RAS, MAPK1/ERK2, MAPK3/ERK1 and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. FGFR2 signaling is down-regulated by ubiquitination, internalization and degradation. Mutations that lead to constitutive kinase activation or impair normal FGFR2 maturation, internalization and degradation lead to aberrant signaling. Over-expressed FGFR2 promotes activation of STAT1. Subunit: Monomer. Homodimer after ligand binding. Interacts predominantly with FGF1 and FGF2, but can also interact with FGF3, FGF4, FGF6, FGF7, FGF8, FGF9, FGF10, FGF17, FGF18 and FGF22 (in vitro). Ligand specificity is determined by tissue-specific expression of isoforms, and differences in the third Ig-like domain are crucial for ligand specificity. Isoform 1 has high affinity for FGF1 and FGF2, but low affinity for FGF7. Isoform 3 has high affinity for FGF1 and FGF7, and has much higher affinity for FGF7 than isoform 1 (in vitro). Affinity for fibroblast growth factors (FGFs) is increased by heparan sulfate glycosaminoglycans that function as coreceptors. Likewise, KLB increases the affinity for FGF19 and FGF21. Interacts with PLCG1, GRB2 and PAK4. Subcellular Location: Cell membrane; Single-pass type I membrane protein. Golgi apparatus. Cytoplasmic vesicle. Note=Detected on osteoblast plasma membrane lipid rafts. After ligand binding, the activated receptor is rapidly internalized and degraded. Isoform 1: Cell membrane; Single-pass type I membrane protein. Note=After ligand binding, the activated receptor is rapidly internalized and degraded. Isoform 3: Cell membrane; Single-pass type I membrane protein. Note=After ligand binding, the activated receptor is rapidly internalized and degraded. Post-translational modifications: N-glycosylated in the endoplasmic reticulum. The N-glycan chains undergo further maturation to an Endo H-resistant form in the Golgi apparatus. Ubiquitinated. FGFR2 is rapidly ubiquitinated after autophosphorylation, leading to internalization and degradation. Subject to degradation both in lysosomes and by the proteasome. DISEASE: Defects in FGFR2 are the cause of Crouzon syndrome (CS) [MIM:123500]; also called craniofacial dysostosis type I (CFD1). CS is an autosomal dominant syndrome characterized by craniosynostosis (premature fusion of the skull sutures), hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism. Defects in FGFR2 are a cause of Jackson-Weiss syndrome (JWS) [MIM:123150]. JWS is an autosomal dominant craniosynostosis syndrome characterized by craniofacial abnormalities and abnormality of the feet: broad great toes with medial deviation and tarsal-metatarsal coalescence. Defects in FGFR2 are a cause of Apert syndrome (APRS) [MIM:101200]; also known as acrocephalosyndactyly type 1 (ACS1). APRS is a syndrome characterized by facio-cranio-synostosis, osseous and membranous syndactyly of the four extremities, and midface hypoplasia. The craniosynostosis is bicoronal and results in acrocephaly of brachysphenocephalic type. Syndactyly of the fingers and toes may be total (mitten hands and sock feet) or partial affecting the second, third, and fourth digits. Intellectual deficit is frequent and often severe, usually being associated with cerebral malformations. Defects in FGFR2 are a cause of Pfeiffer syndrome (PS) [MIM:101600]; also known as acrocephalosyndactyly type V (ACS5). PS is characterized by craniosynostosis (premature fusion of the skull sutures) with deviation and enlargement of the thumbs and great toes, brachymesophalangy, with phalangeal ankylosis and a varying degree of soft tissue syndactyly. Three subtypes of Pfeiffer syndrome have been described: mild autosomal dominant form (type 1); cloverleaf skull, elbow ankylosis, early death, sporadic (type 2); craniosynostosis, early demise, sporadic (type 3). Defects in FGFR2 are the cause of Beare-Stevenson cutis gyrata syndrome (BSCGS) [MIM:123790]. BSCGS is an autosomal dominant condition is characterized by the furrowed skin disorder of cutis gyrata, acanthosis nigricans, craniosynostosis, craniofacial dysmorphism, digital anomalies, umbilical and anogenital abnormalities and early death. Defects in FGFR2 are the cause of familial scaphocephaly syndrome (FSPC) [MIM:609579]; also known as scaphocephaly with maxillary retrusion and mental retardation. FSPC is an autosomal dominant craniosynostosis syndrome characterized by scaphocephaly, macrocephaly, hypertelorism, maxillary retrusion, and mild intellectual disability. Scaphocephaly is the most common of the craniosynostosis conditions and is characterized by a long, narrow head. It is due to premature fusion of the sagittal suture or from external deformation. Defects in FGFR2 are a cause of lacrimo-auriculo-dento-digital syndrome (LADDS) [MIM:149730]; also known as Levy-Hollister syndrome. LADDS is a form of ectodermal dysplasia, a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. LADDS is an autosomal dominant syndrome characterized by aplastic/hypoplastic lacrimal and salivary glands and ducts, cup-shaped ears, hearing loss, hypodontia and enamel hypoplasia, and distal limb segments anomalies. In addition to these cardinal features, facial dysmorphism, malformations of the kidney and respiratory system and abnormal genitalia have been reported. Craniosynostosis and severe syndactyly are not observed. Defects in FGFR2 are the cause of Antley-Bixler syndrome (ABS) [MIM:207410]. ABS is a multiple congenital anomaly syndrome characterized by craniosynostosis, radiohumeral synostosis, midface hypoplasia, malformed ears, arachnodactyly and multiple joint contractures. ABS is a heterogeneous disorder and occurs with and without abnormal genitalia in both sexes. Similarity: Belongs to the protein kinase superfamily. Tyr protein kinase family. Fibroblast growth factor receptor subfamily. Contains 3 Ig-like C2-type (immunoglobulin-like) domains. Contains 1 protein kinase domain. SWISS: P21802 Gene ID: 2263 Database links: Entrez Gene: 2263 Human Entrez Gene: 14183 Mouse Entrez Gene: 25022 Rat Omim: 176943 Human SwissProt: P21802 Human SwissProt: P21803 Mouse Unigene: 533683 Human Unigene: 16340 Mouse Unigene: 12732 Rat Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. KGFR又称FGFR2(Fibroblast Growth Factor Receptor 2)成纤维细胞生长因子受体又称:纤维母细胞生长因子受体2,是FGFRs家族的一员。不同的FGFR对FGF亲和力不同,在组织的分布也不一样。FGFR-2对细胞的增殖、分化、血管生成、胚胎及骨骼发育和在与生长发育相关的进程中起着十分重要的作用. |
| 产品图片 |  Paraformaldehyde-fixed, paraffin embedded (mouse brain tissue); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. Paraformaldehyde-fixed, paraffin embedded (mouse brain tissue); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining.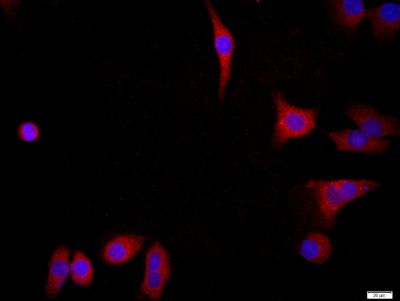 Tissue/cell: MCF7; 4% Paraformaldehyde-fixed; Triton X-100 at room temperature for 20 min; Blocking buffer (normal goat serum, C-0005) at 37°C for 20 min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) 1:200, 90 minutes at 37°C; followed by a conjugated Goat Anti-Rabbit IgG antibody (bs-0295G-FITC) at 37°C for 90 minutes, DAPI (blue, C02-04002) was used to stain the cell nuclei.Image was kindly submitted by Dr. Uthaman from Yale University. L6 cells were transfected with FGFR2, and stained with RABBIT ANTI-FGFR2 POLYCLONAL ANTIBODY, conjugated (bs-0675R-FITC) at 1:100 dilution Tissue/cell: MCF7; 4% Paraformaldehyde-fixed; Triton X-100 at room temperature for 20 min; Blocking buffer (normal goat serum, C-0005) at 37°C for 20 min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (bs-0675R) 1:200, 90 minutes at 37°C; followed by a conjugated Goat Anti-Rabbit IgG antibody (bs-0295G-FITC) at 37°C for 90 minutes, DAPI (blue, C02-04002) was used to stain the cell nuclei.Image was kindly submitted by Dr. Uthaman from Yale University. L6 cells were transfected with FGFR2, and stained with RABBIT ANTI-FGFR2 POLYCLONAL ANTIBODY, conjugated (bs-0675R-FITC) at 1:100 dilution |





